Indirect Referencing
When acquiring X nucleus spectrum such as 11B, 31P or 29Si, the spectrum must be referenced to something other than the solvent, since the solvent likely does not contain the nucleus. Either an internal standard or indirect referencing can be used. Sometimes, internal standards will react with your compound, or the correct internal standard compound can be difficult to obtain. Indirect referencing can provide accurate referencing without having to add an internal standard. The following instructions are for indirect referencing in Topspin (NEO400, AVII600, AVIII600, AV400). Scroll down for indirect referencing in MNova.
Indirect Referencing in Topspin
First, acquire both a 1H and X nucleus 1D spectrum, as normal.
Try the Indirect Referencing Calculator on Google Docs!
Calibrating Data:
- For calibrating 1H data, calibrate to the TMS/DSS peak (if using an internal standard), or the residual solvent peak.
- To calibrate, zoom in to the general area of the peak you want to calibrate.
- Click the Calib. Axis button or type .cal
- The cursor will turn red, and then click on the center of the solvent peak.
- Enter the corrected chemical shift in the pop-up box.
Indirect Referencing for X Nuclei:
- Acquire and calibrate a proton spectrum as directed above
- In the proton spectrum, type s sf to get the spectrometer frequency for 1H (should be 600.39000XX on the AVIII600)
- Find the constant for the Frequency Ratio Ξ in the table below.
- Equation to calculate the indirect referencing constant:
600.39000XX (from step b)/100*Freq Ratio from table below
- Go to the X nucleus spectrum (11B, 29Si, etc.) and type s sf
- Enter the value calculated in step (d)
- Now your X nucleus spectrum is calibrated without having to use an external or internal standard.
Reference: Nuclear spin properties and conventions for chemical shifts. Harris, et al. 2001 (PDF)
| Isotope | Freq Ratio | Isotope | Freq Ratio |
|---|---|---|---|
| 1H | 100 | 51V | 26.302948 |
| 2H | 15.350609 | 69Ga | 24.001354 |
| 7Li | 38.863797 | 71Ga | 30.496704 |
| 11B | 32.083974 | 77Se | 19.071513 |
| 13C* | 25.145004 | 79Br | 25.054454 |
| 13C** | 25.145022 | 87Rb | 32.720454 |
| 14N | 7.226330 | 89Y | 4.000198 |
| 15N | 10.136767 | 91Zr | 9.296298 |
| 17O | 13.556457 | 103Rh | 3.186447 |
| 19F | 94.094008 | 109Ag | 4.653533 |
| 23Na | 26.451921 | 113Cd | 22.193175 |
| 27Al | 26.056859 | 119Sn | 37.290632 |
| 29Si | 19.867187 | 171Yb | 17.499306 |
| 31P | 40.480742 | 183W | 4.166387 |
| 35Cl | 9.797909 | 195Pt | 21.496784 |
| 45Sc | 24.291702 | 207Pb | 20.920599 |
*Neat TMS scale =1% TMS CDCl3 13C chemical shifts + 0.71 ppm, Adamantane = 38.48 ppm and 1.82 ppm
**1 % TMS in CDCl3, adamantane = 37.77 ppm and 1.71 ppm
Indirect Referencing in MNova
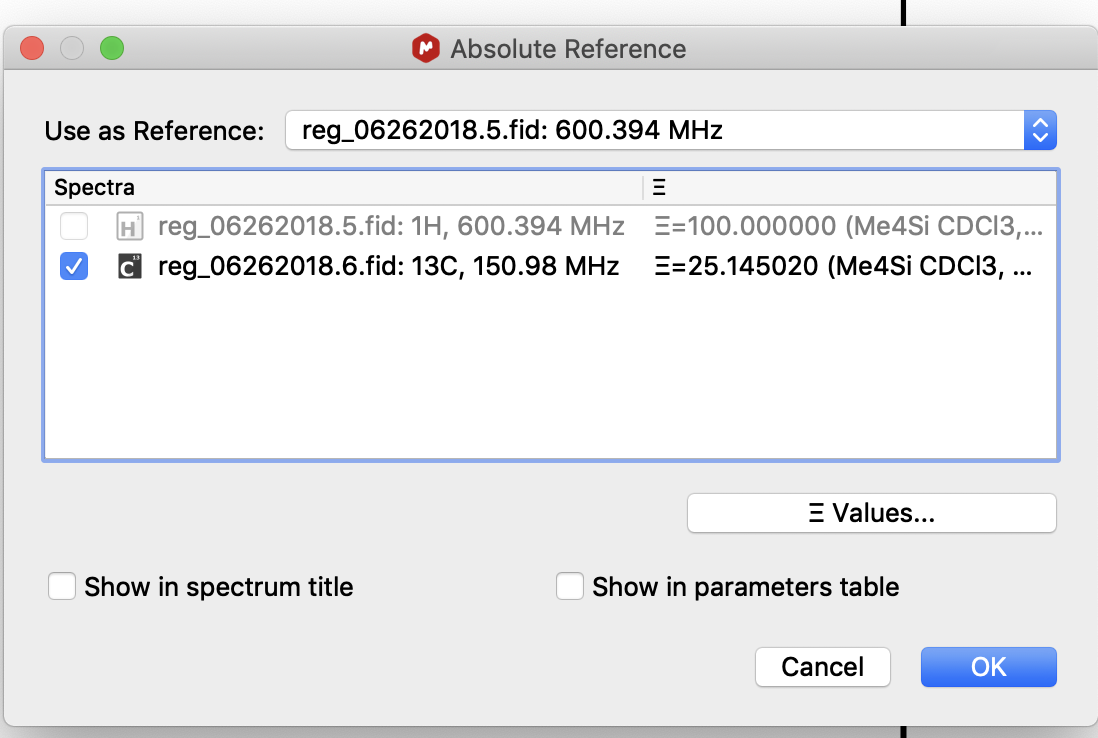
1. Acquire a 1H and the X nucleus spectrum of your choosing (13C, 31P, 29Si, 11B, etc.)
2. Open the 1H and X nucleus spectra in MNova.
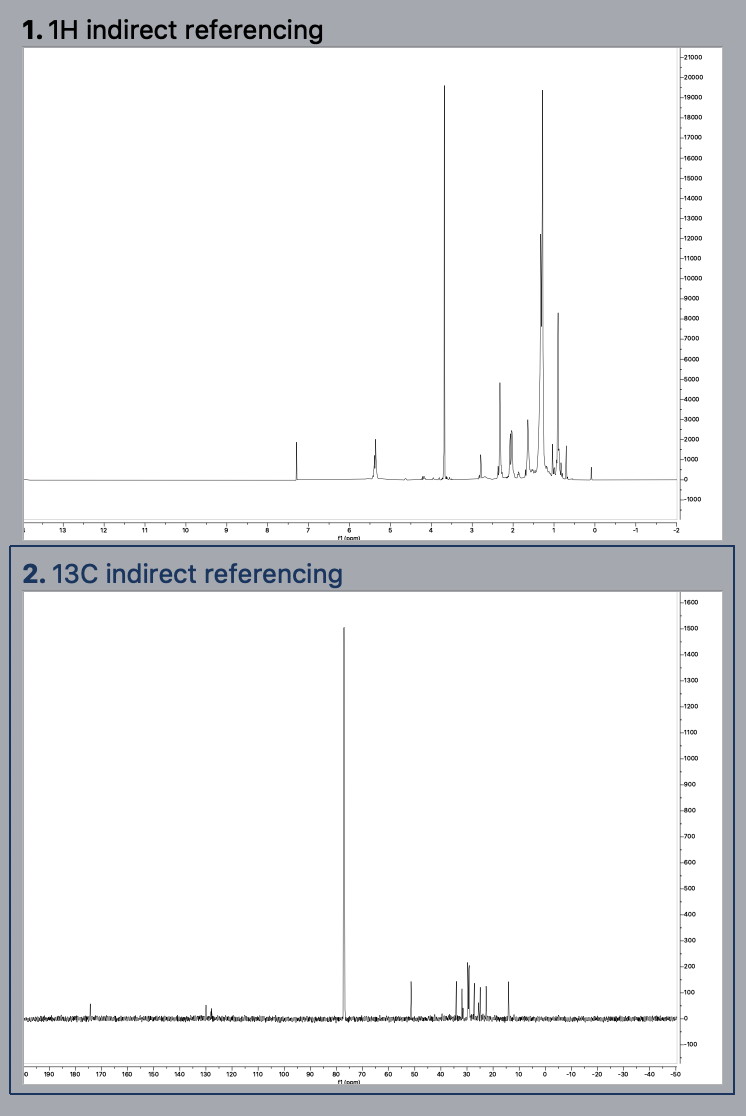
3. Calibrate the solvent peak or TMS peak in the 1H spectrum.
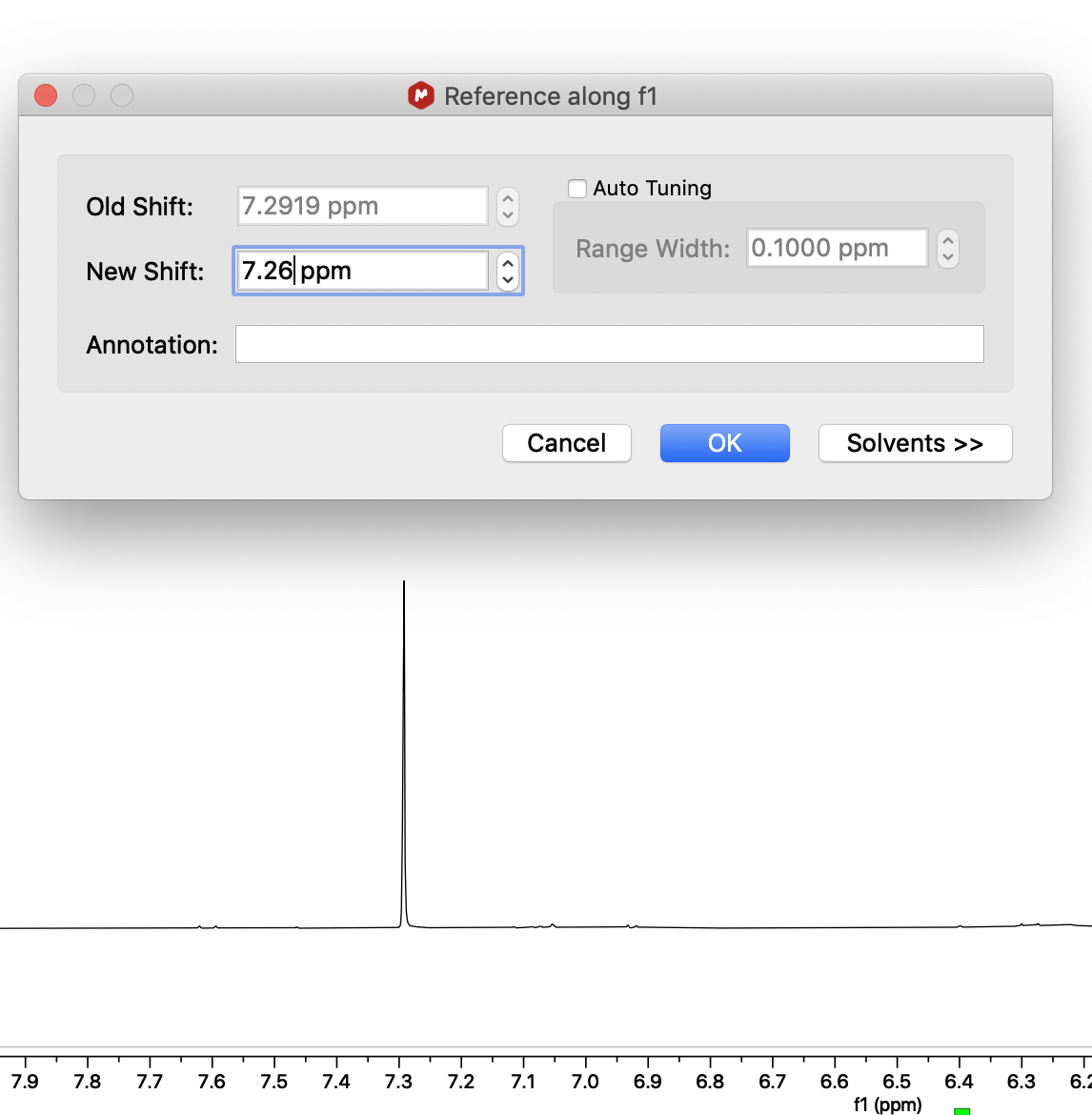
4. Go to the X nucleus spectrum. Click the button next to the reference button and select "Absolute Reference..."
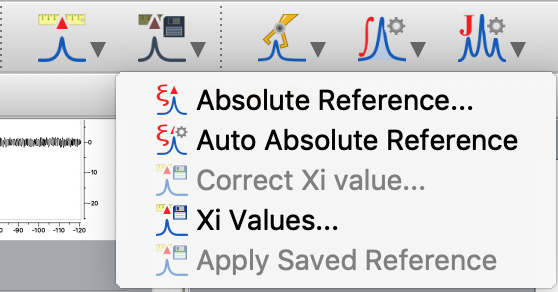
5. Make sure the calibrated 1H is selected in the drop-down box. Tick the check marks next to the spectra you wish to calibrate relative to the 1H spectrum. (In this example, we only have on other spectrum open, but in theory, you could have several spectra you are calibrating relative to the same 1H.) Click OK. Now your X nucleus spectrum is indirectly referenced to the chosen 1H spectrum.