Mass Spectrometry Tutorial (Dr. Kamel Harrata)
This tutorial discusses basic aspects of mass spectrometry that will be helpful to you in deciding the proper techniques and measurements for your research samples.
- What is Mass Spectrometry?
- The Mass Spectrometer
- Ionization Methods
- Analysis of Ions
- Tandem Mass Spectrometry
- Separation Methods for Coupling with Mass Spec
Mass spectrometry is an analytical technique that involves the study in the gas phase of ionized molecules with the aim of one or more of the following:
- Molecular weight determination
- Structural characterization
- Gas phase reactivity study
- Qualitative and quantitative analysis of components in a mixture.
Mass spectrometry consists basically of weighing ions in the gas phase. The instrument used could be considered as a sophisticated balance which determines with high precision the masses of individual atoms and molecules. Depending on the samples chemical and physical properties, different ionization techniques can be used. One of the main factor in choosing which ionization technique to be used is thermolability. For samples that are not themolabile and relatively volatile, ionization such as Electron Impact and/or Chemical Ionization can be effectively used. For samples that are thermolabile such as peptides, proteins and other samples of biological interest, soft ionization techniques are to be considered. Among the most used soft ionization techniques are Electrospray (ESI) and Matrix Assisted Laser Desorption (MALDI). The name given to a particular mass spec technique is usually pointing to the ionization method being used.
Atomic and molecular masses are assigned relative to the mass of the carbon isotope, 12C, whose atomic weight is defined as exactly 12. The actual mass of 12C is 12 daltons, with one dalton is equal to 1.661 10-24 g. The mass of a molecule or an ion can be presented in daltons (Da) or kilodaltons (kDa).
Mass spectrometry uses an instrument called a mass spectrometer. The main components of a mass spectrometer are:
- Inlet system (LC, GC, Direct probe etc...)
- Ion source (EI, CI, ESI, APCI, MALDI, etc...)
- Mass analyzer (Quadrupole, TOF, Ion Trap, Magnetic Sector)
- Detector (Electron Multiplier, Micro Channel Plates MCPs)
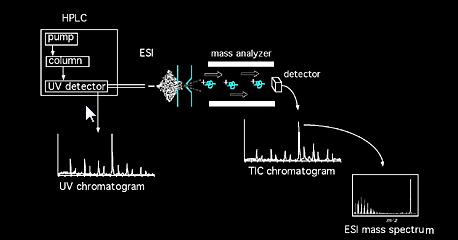
Samples can be introduced to the mass spectrometer directly via solids probe, or in the case of mixtures, by the intermediary of chromatography device (e.g. Gas chromatography, Liquid chromatography, Capillary electrophoresis, etc...). Once in the source, sample molecules are subjected to ionization. Ions formed in the source (molecular and fragment ions) acquire some kinetic energy and leave the source. A calibrated analyzer then analyzes the passing ions as a function of their mass to charge ratios. Different kind of analyzer(s) can be used, Magnetic, Quadrulpole, Ion trap, Fourier Transform, Time of Flight, etc...The ion beam exiting the analyzer assembly is then detected and the signal is registered. Common ionization method acronyms include:
- EI=Electron Impact;
- CI=Chemical Ionization;
- SIMS=Secondary Ions Mass Spec;
- FAB=Fast Atom Bombardment;
- LDMS=Laser Desorption Mass Spec;
- PDMS=Plasma Desorption Mass Spec;
- TS=Thermospray;
- AS=Aerospray;
- ESMS=Electrospray Mass Spec.
Common mass analyzer acronyms include:
- EB=Electrostatic-Magnetic;
- IT=ion trap;
- Q=Quadrupole;
- TOF=Time of Flight.
Selection of the proper ionization method for the analysis of your sample is extremely important. Although we can offer suggestions, it is your responsibility to understand and select the method(s) appropriate for your research compounds.
- Electron Impact EI Ionization
- Chemical Ionization CI
- Negative Ion Chemical Ionization
- Electrospray Ionization Techniques
- Matrix Assisted Lazer Desorption (not offered in our facility, but available elswhere on campus)
- Atmospheric Pressure Chemical Ionization APCI
M + e-(70eV) -----> M+. + 2e-
EI ionization method is suitable for non thermolabile compounds. The volatility of the sample is required. Sample molecules in vapor state are bombarded by fast moving electrons, conventionally 70 eV energy. This results in ion formation. One electron from the highest orbital energy is dislodged, and as a consequence molecular ions are formed. Some of this molecular ions decompose and fragment ions are formed. The fragmentation of a given ion is due to the excess of energy that it requires within the ionization. Fragment ions can be odd electron or even electron. Molecular ions formed in electron impact ionization are odd electron ions. Odd electron fragment ions are formed by direct cleavage(e.g. direct cleavage of a C-C bond). Even electron fragment ions are often formed by rearrangement(e.g. proton transfer). Sample can be introduced to the EI source via a gas chromatography device, for example in the case of mixtures, or directly via a solids probe device. The quantities needed for an experiment is usually less than a microgram of material.
EI mass spectra, in most of cases, contain intense fragment ion peaks and much less intense molecular ion peak. When the molecular ion peak is not observed in the mass spectrum, chemical ionization can be used in order to get molecular ion information. One helpful rule for determining whether an ion is a molecular ion is the Nitrogen Rule.
Nitrogen rule: As indicated above, molecular ions formed in EI ionization are odd electron ions. If their observed mass to charge ratio is odd, the molecule under investigation contains an odd number of nitrogen atoms. If that mass to charge ratio is an even number, that molecule contains no or even Nitrogen atoms.
For organic chemists, Chemical Ionization (CI) is especially useful technique when no molecular ion is observed in EI mass spectrum, and also in the case of confirming the mass to charge ratio of the molecular ion. Chemical ionization technique uses virtually the same ion source device as in electron impact, except, CI uses tight ion source, and reagent gas. Reagent gas (e.g. ammonia) is first subjected to electron impact. Sample ions are formed by the interaction of reagent gas ions and sample molecules. This phenomenon is called ion-molecule reactions. Reagent gas molecules are present in the ratio of about 100:1 with respect to sample molecules. Positive ions and negative ions are formed in the CI process. Depending on the setup of the instrument (source voltages, detector, etc...) only positive ions or only negative ions are recorded.
In CI, ion molecule reactions occur between ionized reagent gas molecules (G) and volatile analyte neutral molecules (M) to produce analyte ions. Pseudo-molecular ion MH+ (positive ion mode) or [M-H]- (negative ion mode) are often observed. Unlike molecular ions obtained in EI method, MH+ and [M-H]- detection occurs in high yield and less fragment ions are observed.
Positive ion mode:
GH+ + M ------> MH+ + G
Negative ion mode:
[G-H]- + M ------> [M-H]- + G
These simple proton transfer reactions are true gas-phase Acid-Base processes in the Bronsted-Lowrey sense. A"tight" ion source (pressure=0.1-2 torr) is used to maximize collisions which results in increasing sensitivity. To take place these ion molecule reactions must be exothermic. Proton transfer is one of the simple processes observed in positive CI:
RH+ + M -----> MH+ + R
One of the decisive parameter in this reaction is the proton affinity. For the reaction to occur, the proton affinity of the molecule M must be higher that the one of the gas molecule. The main reagent gases used in CI are: Ammonia, Methane, and Isobutane. The predominant reactant ions formed are given in the mechanisms shown below. Choice of reagent gas affects the extent of fragmentation of the quasi-molecular ion.
Methane (positive ion chemical ionization):
- CH4 + e -----> CH4+. + 2e ------> CH3+ + H.
- CH4+. + CH4 -----> CH5+ +CH3.
- CH4+. + CH4 -----> C2H5+ + H2 + H.
Isobutane (positive ion chemical ionization):
- i-C4H10 + e -----> i-C4H10+. + 2e
- i-C4H10+. + i-C4H10 ------> i-C4H9+ + C4H9 +H2
Ammonia (positive ion chemical ionization):
- NH3 + e -----> NH3+. + 2e
- NH3+. + NH3 ------> NH4+ + NH2.
- NH4+ + NH3 --------->N2H7+
In methane positive ion mode chemical ionization the relevant sample peaks observed are MH+, [M+CH5]+, and [M+C2H5]+; but mainly MH+. This corresponds to the masses M+1, M+29, and M+41.
In isobutane positive ion mode chemical ionization the main peak observed is MH+.
In ammonia positive ion mode chemical ionization the main peaks observed are MH+, and [M+NH4]+. If more than one protonation site is present, additional NH3 adducts might be seen corresponding to [M+NH3+NH4]+. This corresponds to the masses M+1, M+18, and M+35.
In some cases, protonated dimers or other adducts might be seen; loss of H2O followed by protonation or adduct ion formation is seen for some classes of compounds. If the spectrum you observe does not seem to show the proper adduct ions, or shows extensive fragmentation, be wary when you try to interpret the results. There is an abundance of data available in the literature discussing chemical ionization mechanisms applicable to specific classes of compounds.
Two factors determine the choice of the reagant gas to be used:
- Proton affinity PA
- Energy transfer
NH3 (ammonia) is the most used reagent gas in CI because of the low energy transfer of NH4+ compare to CH5+ for example. With NH3 as reagent gas, usually MH+ and MNH4+ (17 mass units difference) are observed.
Negative Ion Chemical Ionization
Three mechanisms can be underlined:
- Electron capture reaction due to attainment of slow moving, low energy "thermalized" electrons which may be transfered more efficiently to sample molecules.
- Electron transfer from ionized reagent gas (e.g. NH2- may transfer an electron to a molecule having a greater electron affinity than NH2).
- Reagent gas ions participate in true CI reactions (e.g. proton abstraction, according to relative acidities).
Molecular ions observed in negative ion chemical ionization mass spectra are usually M- or [M-H]-.
Electrospray Ionization Method
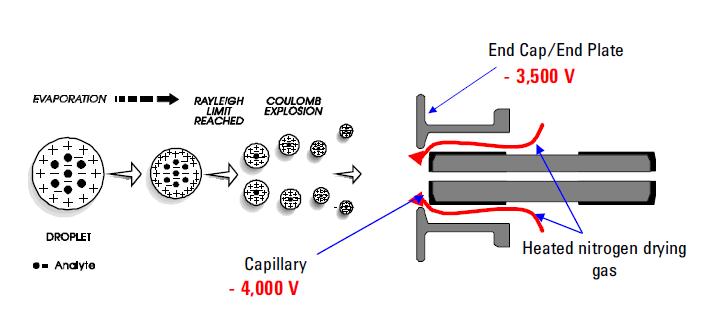
Among the most used spray ionization techniques is Electrospray Ionization (ESI). This technique continues to be the method of choice for analyzing thermolabile chemicals. Its capabilities are well documented. It uses an electrical stress between the ESI probe exit (e.g. capillary) and the counter electrode, which is located few millimeters from the probe. The process results in the generation of highly charged droplets directly from the infused solution. Multiply and/or singly charged analyte molecules desorbe from the sprayed droplets and sampled through the rest of the mass spectrometer. ESI has been distinguished for its ability to produce multiply charged molecular ions from a large variety of polymers such as protein and DNA fragments; it allows also sensitive detection of singly charged low molecular weight polar species such as drugs and drug metabolites. The formation of positive or negative ions (depending on the sign of the applied electrical field) occurs in high yield. In the positive ion mode protonated and/or alkali adduct analyte molecules generally observed in the mass spectra. In the negative ion mode operation peaks corresponding to deprotonated analyte molecules are observed. ESI is described as a very "soft" ionization technique where the surrounding bath gas has a moderating effect on the internal and translational energies of desorbed ions.
Advantages of ESI:
- Soft ionization process so intact molecular ions are observed
- ESI allows production of multiply charged ions. This results in the ability of analyzing very high molecular weight species using the most available mass analyzers (e.g. quadrupoles).
- ESI is an atmospheric pressure process. This makes it easy to use and easy to interface with HPLC and CE separation techniques.
Matrix Assisted Laser Desorption (MALDI)
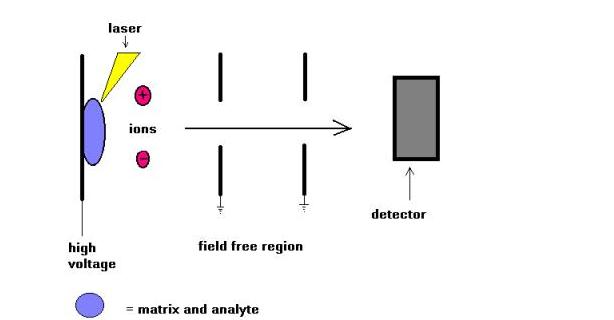
Matrix Assisted Laser Desorption (MALDI) mass spectrometry technique was introduced by Karas and Hillkamp in 1988 for the ionization of peptides and proteins. Soon there after this technique was able to analyze other type of biomolecules, such as oligosaccharides, glycolipids, nucleotides, and synthetic polymers. In this technique, samples are cocrystallized with a UV-absorbing substance called matrix. For example for proteins, the matrix of choice is often sinapinic acid. A 337 nm radiation from nitrogen laser is most commonly used. The laser helps introducing energy into the molecular system in such a way preventing thermal decomposition.
MALDI is often used with time-of-flights mass spectrometers ( TOF ) due to the pulsing nature of the technique, and the mass range capability. Molecular weights up to few hundreds of daltons could be measured. Comparison of MALDI and ESI ionization techniques was attempted in the last few years. In my opinion these two techniques are not competitive but complementary. Just to name a few, for high molecular weight species, MALDI leads to the formation of singly charged molecular ions while ESI allows the formation of multiply charged molecular ions.
Practical considerations:
- The final molar ratio sample/matrix is about or around 1/5000.
- Final concentration of the sample is from 1 to 10 pmol/ul
- Our experience with MALDI point to a dynamic range of 100 fmol/ul to few hundreds pmol/ul
- MALDI is relatively robust ionization technique that tolerates the use of salts and surfactants and buffers. Although it is best to remove them for better performance.
Peptide and Protein Standards for MALDI:
- Angiotensin II (human) MW: 1046.2
- Substance P (human) MW: 1347.7
- Insulin (bovine) MW: 5733.6
- Cytochrom c (equine) MW: 12,360.1
- RNase A (bovine) MW: 13,682.2
- Apo-Myoglobin (equine) MW: 16,951.5
- Trypsinogen (bovine) MW: 23,980.9
Atmospheric Pressure Chemical Ionization
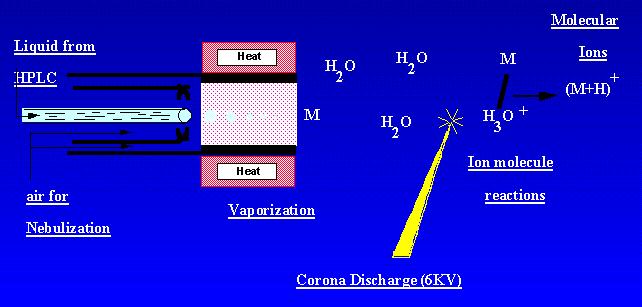
APCI is a technique which creates ions at atmospheric pressure. A sample solution flows through a heated tube where it is volatilized in a mist and sprayed into a corona discharge with the aid of nitrogen nebulization. Sample molecular are ionized by ion molecule reactions from the ambiant corona discharge ions. Ions are produced in the discharge and extracted into the mass spectrometer. APCI is best suited to relatively polar, semi-volatile samples. An APCI mass spectrum usually contained the quasi-molecular ion, [M-H]- or [M + H]+.
It is possible to use several different physical parameters to achive mass seperation. Common types of mass analyzers are discussed below. Each has advantages and disadvantages. In our facility we currently have Quadrupole, Ion Trap, amd Time-of-Flight (TOF) mass spectrometers.
Magnetic Sector Mass Spectrometer
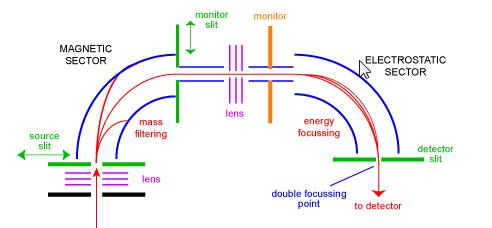
The sector mass spectrometer was one of the most common types of mass analyser and probably the most familiar to the everyday scientist. In the 1950's, the first commercial mass spectrometers were sector instruments. They consist of some combination of a large electromagnetic, and some kind of electrostatic focussing device. Different manufactures use differing geometries. Figure 1 shows a schematic of a standard 'BE' geometry double focussing instrument. The BE configuration is also called reverse geometry sector mass spectrometer - that is, a dual sector instrument consisting of a magnetic sector followed by an electrostatic sector.
Ions enter the instrument from the source (bottom left) where they are initially focussed. They enter the magnetic sector through the source slit where they are deflected according to the left-hand rule. Higher-mass ions are deflected less than lower-mass ions. Scanning the magnet enables ions of different masses to be focussed on the monitor slit. At this stage, the ions have been separated only by their masses. To obtain a spectrum of good resolution where all ions with the same m/z appear coincident as one peak in the spectrum, ions have to be filtered by their kinetic energies. After another stage of focussing the ions enter the electrostatic sector where ions of the same m/z have their energy distributions corrected for and are focussed at the double focussing point on the detector slit.
Sector instruments had huge commercial successes in the 1950's and 1960's as they were the only practical way of obtaining high-resolution data. In the last 20 years or so, with the decreasing prices of FTMS and the development of high-resolution alternatives (for example Q-Tof) sector instruments are in decline.
Time-of-Flight Mass Spectrometry (TOF-MS)
A time-of-flight mass spectrometer uses the differences in transit time through a drift region to separate ions of different masses. It operates in a pulsed mode so ions must be produced or extracted in pulses. An electric field accelerates all ions into a field-free drift region with a kinetic energy of qV, where q is the ion charge and V is the applied voltage. Since the ion kinetic energy is 0.5mv2, lighter ions have a higher velocity than heavier ions and reach the detector at the end of the drift region sooner.
Theory:
- K.E. = qV
- 1/2 mv2 = qV
- v = (2qV/m)1/2
The transit time (t) through the drift tube is L/v where L is the length of the drift tube
- t=L / (2V/m/q)1/2
Schematic of a linear TOF-MS
This schematic shows ablation of ions from a solid sample with a pulsed laser. The reflectron is a series of rings or grids that act as an ion mirror. This mirror compensates for the spread in kinetic energies of the ions as they enter the drift region and improves the resolution of the instrument. The output of an ion detector is displayed on an oscilloscope as a function of time to produce the mass spectrum.
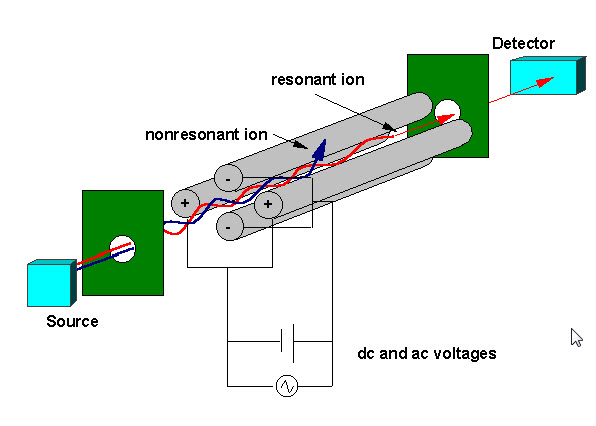
Ions created by electron impact (EI), electrospray (ESI), or matrix-assisted laser desorption (MALDI) ionization are focused using an electrostatic lensing system into the ion trap. An electrostatic ion gate pulses open (-V) and closed (+V) to inject ions into the ion trap. The pulsing of the ion gate differentiates ion traps from "beam" instruments such as quadrupoles where ions continually enter the mass analyzer. The time during which ions are allowed into the trap, termed the "ionization duration", is set to maximize signal while minimizing space-charge effects. Space-charge results from too many ions in the trap that cause a distortion of the electrical fields leading to an overall reduction in performance. The ion trap is typically filled with helium to a pressure of about 1 mtorr. Collisions with helium dampens the kinetic energy of the ions and serve to quickly contract trajectories toward the center of the ion trap, enabling trapping of injected ions. Trapped ions are further focused toward the center of the trap through the use of an oscillating potential, called the fundamental rf , applied to the ring electrode. An ion will be stably trapped depending upon the values for the mass and charge of the ion, the size of the ion trap (r), the oscillating frequency of the fundamental rf ( w), and the amplitude of the voltage on the ring electrode ( V). The dependence of ion motion on these parameters is described by the dimensionless parameter qz, qz = 4eV/mr2w2
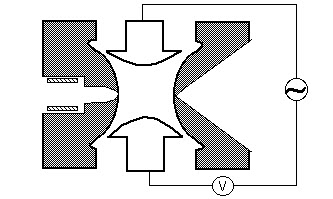
Quadrupole
A quadrupole mass filter consists of four parallel metal rods arranged as in the figure below. Two opposite rods have an applied potential of (U+Vcos(wt)) and the other two rods have a potential of -(U+Vcos(wt)), where U is a dc voltage and Vcos (wt) is an ac voltage. The applied voltages affect the trajectory of ions traveling down the flight path centered between the four rods. For given dc and ac voltages, only ions of a certain mass-to-charge ratio pass through the quadrupole filter and all other ions are thrown out of their original path. A mass spectrum is obtained by monitoring the ions passing through the quadrupole filter as the voltages on the rods are varied. There are two methods: varying w and holding U and V constant, or varying U and V (U/V) fixed for a constant w.
Tandem Mass Spectrometry, usually referred to as MS/MS, involves the use of 2 or more mass analyzers. It is often used to analyze individual components in a mixture. This technique adds specificity to a given analysis. Although tandem mass spectrometry can be referred to MS/MS, MS/MS/MS, etc..., in this presentation I am going to describe only MS/MS.
The basic idea of MS/MS is a selection of a m/z of a given ion formed in the ion source, and subject this ion to fragmentation, usually by collision with inert gas (eg. Argon). The product ions are then detected. This is a powerful way of confirming the identity of certain compounds and determining the structure of unknown species. So MS/MS is a process that involves 3 steps: ionization,mass selection,mass analysis.
MS/MS could be performed on instruments such as triple quadrupole (QQQ), ion trap, time of flight, fourier transform, etc... The triple quadrupole is the most frequently used mass spectrometer for MS/MS, perhaps because of the cost and ease of use among other factors.
Separation Methods for Coupling with Mass Spec
- GC-MS: Sample mixtures are directly vaporized and enter bonded fused silica columns. Components of the mixture are separated based on their affinity difference with the bonded phase. Separated compounds exit the column and enter the vacuum system of the mass spectrometer. Sample molecules are ionized (EI, or CI), and accelerated into a precalibrated mass analyzer (e.g. Q, Ion Trap, TOF, FTMS etc...). Retention times, molecular masses, and fragmentation patterns are recorded. One of the most important considirations of GC/MS is that the sample(s) must be non thermolable meaning thermally stable.
- LC-MS: For compounds that are thermally unstable, LC/MS method is considered. The separation is based on the diffrence in affinity of samples with stationary phase and the mobile phase. e.g. hydrophobicity in case of RP chromatography.
- CZE-MS: This method is based on diffrences of electrophoretic mobility of samples when the fused silica column is under a potential difference between injection side and detector side.
- CIEF-MS: This is a variant of CZE. It is based on diffrerences in isoelectric points of analytes.